Low-abundance analysis in proteomics is billion-dollar valuable but unsolved until now. After 18 months in stealth mode, we announce SorcererScore(tm), a breakthrough in analytics that successfully finds low-abundance, modified peptides (LAMPs).
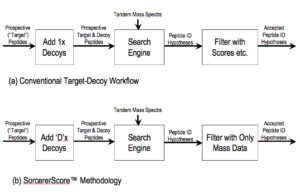
Figure 1: Peptide Identification Part of Proteomics Workflow
Deep proteomics needs both high-accuracy mass data and quality interpretation. Many proteomics labs produce the former but struggle with the latter, akin to an x-ray lab that produces super-sharp images but grossly mis-identifies early tumors. Funding and attention will explode once quality results can be dependably delivered.
The issue is analytics, defined as the discovery of meaningful patterns in data using math and computing. Every big-data field invests heavily in beefy server-based analytics to data-mine deep insights, but not proteomics. This fact alone should raise red flags over popular speedy PC programs. The problem is depth perception: many are unable to see beyond the shallow in judging tools and expertise. This stalls the field but presents opportunity.
How the Emperor lost his clothes
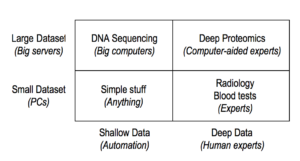
Many factors caused the proteomics community as a whole to distrust professional tech developers, viewed by some as over-priced charlatans, resulting in shoddy digital construction on weak foundations. These include confusing data depth with data volume (see table 1), over-reliance on the false discovery error rate (FDR), and the trivialization of math and computing as ad hoc “programming” by beginners.
Table 1: Analysis Need vs. Data Characteristics
Probably the real factor is culture clash. In my experience, a lot of analytics talent are gifted mathematicians and engineers who, like those gifted in basketball or comedy, take pride in being self-taught “artists” who eschew advanced degrees. The tech culture confuses the medical community used to judging people only by institutional credentials. Bridging the medicine-technology divide is likely the greatest opportunity of our lifetime.
In any case, labs should not be surprised when they spend lavishly on mass spectrometers but next to nothing on analytics, only to get beautiful data but questionable results. The No-Free-Lunch principle applies. Anyone who thinks he’s getting something for nothing — cheap great software, all-you-can-consume cloud computing, etc. — is simply not understanding the full trade-off.
One common mistake is to over-generalize from a small selected sample. A peer-reviewed program that worked on two datasets, like a compound that cured some rats, may not generalize. Cloud computing, the Airbnb of CPUs, is cost-effective for infrequent use (paying 5x on-demand is only half the cost vs. 10% utilization) but not for continuous data-mining. Inexpensive PC search engines achieve huge speedup using “full-indexing” for already fast search conditions, used in contrived benchmarks against SORCERER’s patented partial indexing, but ironically that technique is useless to speed up slow searches because the indexing operation itself can take days.
The biggest problem with non-robust software is hackability. Such software allows users, even inadvertently, to push algorithms beyond their range of validity to yield better-than-reality results (though some labs seem not to mind). Researchers report getting as much as 50% more peptide IDs by tightening mass tolerances (< 10 ppm) and multiple IDs from each spectrum. Sounds great until one looks deeper. The problem is, common sense suggests it must be rare to have even one pair of peptides with three near-identical parameters of (chromatographic) retention time, charge and mass. For the software to report a sizable percentage of these should be a cause for concern. The weak link is the fragile FDR estimation susceptible to aggressive decoy-filtering with over-tight mass tolerances. The latter shrinks the correct-vs-wrong mass-error differential (the only information from mass spectrometry), making it easier for any algorithm to mistaken wrong IDs as correct. Professionally developed software, often criticized for not matching the raw ID quantity of low-priced software, is engineered to minimize such miscalculations.
Low-end software sacrifices accuracy for speed by using assumptions to skip processing. For example, Bayesian techniques need less data (and hence less computing) to estimate probabilities, which sounds great until one looks deeper. Fundamentally, they are designed to squeeze the most out of scarce or poor quality data, particularly in data-starved fields like economics, by supplementing them with “a priori” assumptions. A direct non-modeled approach is preferred if both data quantity and quality allow it. For instance, if you want the most accurate estimation of the ‘heads’ probability of a weighted coin, the very best way is brute-force: Just average 100 or 1000 random samples. It’s only if you are unable or unwilling to obtain enough samples that you introduce a priori probabilities. Assumption-riddled software is full of fudge factors tuned to the same cow protein etc. casual users use to “validate” it. When shortcuts are hidden deep within opaque analytics, it is impossible to spot quality problems before it’s too late.
Mathematics helps one cut through the clutter from ad hoc trial-and-error that plagues our field. For instance, once you can express all major search engine scores in terms of a vector dot-product, it becomes obvious that, unless you work in a demo lab with artificially low noise data, you must use a cross-correlation search engine to improve signal-to-noise — and only one hardwired for peptide mass periodicity, not hacked to accommodate fragment mass tolerance. The reason: the dot-product should always be the first major calculation, so a dot-product with a noise-correction term (aka cross-correlation) builds in noise-suppression. In contrast, scores based on counting matched fragment peaks require snapping each fragment peak to ‘1’ or ‘0’ — i.e. count or don’t count — before the dot-product. This implies a noise pre-filter, so the dot-product is no longer the first calculation. Even the X-Tandem hyper-score can be expressed as a dot-product (of the logarithm of the fragment peaks, then exponentiated) without the noise-suppression. Contrary to common misconception, search engines are pretty generic peptide-to-spectrum pattern matchers that do not and should not be tied to specific mass spectrometer technologies.
Why server scripts
After a dozen years in this field, it’s clear high-value proteomics requires a server-based scripting platform — just like basically every other big-data field — to provide robust, transparent, and flexible capability to handle the idiosyncrasies of individual datasets.
A movie script is not just lines on a page but the rich story they weave by describing interactions between actors. Anyone can take some French classes and write a simple French movie script, but it won’t be very good (no offense). A computer script describes a dynamic story through precise interactions between data and functions. (Unlike a movie script, a computer script crashes or miscalculates with one bad typo.) A server script describes interactions among large subsystems, such as the R statistical engine and the relational database, usually requiring different computer languages for each subsystem. The development of a robust server script requires professional developers with analytics expertise. However, it requires less specialized skills to review and to make minor edits. We believe that is the right paradigm for deep proteomics.
An application-specific server appliance like the SORCERER iDA is optimized at a system level, for example by providing abundant RAM for R and disk-space for indexed searches, whose architecture is difficult to replicate in amorphous cloud computing environments.
Most server software development requires specialty consultants and extensive system-wide testing, which makes it 10x costlier than single-language development of PC programs.
Simple & robust on proven technologies
SorcererScore was designed to meet three objectives: (1) optimized for LAMPs, (2) rigorously hypothesis-driven, and (3) simple enough to understand for a high school student. (See here for background.) The result is Figure 1b above.
SorcererScore is built on three foundation technologies — the search engine, target-decoy search, and the pipeline methodology from the labs of Professors John Yates (Scripps), Steven Gygi (Harvard) and Ruedi Aebersold (ETH Zurich), respectively.
The conventional workflow (Figure 1a) was evolved from low-accuracy data. It uses a 1:1 distribution of target and decoy peptide prospects, which are searched by a search engine to enrich to the top scoring “peptide ID hypotheses” (aka peptide-sequence matches). These are filtered using the search score along with other parameters.
SorcererScore (Figure 1b) has a similar structure but with more decoys and a secret-sauce hypothesis filter using only mass data and decoy metadata. Significantly, neither search scores nor any other parameters are used for filtering. In practice, one would search with wide mass tolerance and keep top 10 or more ID hypotheses, each considered independently without priority. Many more decoys (i.e. D >> 1) can be generated by scrambling target sequences and by including non-sense variable modifications (e.g. add +50 amu to leucine, +60 amu to valine, etc.).
The concept is simple to understand when one realizes there are only two fundamental information available: (1) mass data and (2) decoy metadata. That’s it. Therefore, these and only these should be used to filter peptide ID hypotheses. Indeed, the hypothesis-driven Scientific Method is captured by the arrow between the last two blocks. In other words, the search engine is actually your robot scientific assistant who comes up with hopefully good hypotheses, so don’t scrimp on it.
As a thought experiment, imagine we increase D, the distribution ratio of decoy to target sequences, from conventional D=1 to D=1000. We can expect a wrong ID being a non-decoy to be ~0.1% chance. For a 100K spectra dataset with 20K viable correct IDs out of 1M hypotheses (= top 10 per spectrum * 100K spectra), we expect to find non-decoys comprising the 20K correct IDs and 0.001*(1M-20K) = 980 wrong non-decoy IDs, for FDR~5%. If we go to D=10K though, the wrong non-decoys and FDR drop 10x to 98 and ~0.5%, respectively. There are tradeoffs to be sure, including needing unreasonably massive compute power, but conceptually it’s possible to use decoys alone to robustly find LAMPs.
There are two considerations. First, smart mass filtering will cut CPU needs way down. Second, D=10K may be quite viable for targeted searches for only the proteins in the pathways you care about. It all depends on the problem you’re trying to solve, which can be coded into a script.
We will prioritize the release of SorcererScore information, and scripts when available, to in-maintenance SORCERER clients so they can start ground-breaking research on low-abundance peptides. As appreciation for past support, we will make the information available at a delayed time to previous SORCERER users on request. Please email sales@SageNResearch.com to be put on the request list.
We will also make the technology available as a confidential analysis service (i.e. we act as a data analysis-only core lab) for new or existing high-value datasets, either working with the mass spec lab and/or the Principal Investigator. This will initially be targeted for clinical or high-value data with potentially important low-abundance and/or modified peptides. Please email sales@SageNResearch.com for information.
1 Comment
Leave your reply.