Before Copernicus, planetary mathematics was ad hoc and difficult to understand. But his heliocentric abstraction needed only simple math to reveal patterns (circles) that enabled deep insight (gravity).
Proteomics researchers suffer analysis paralysis from today’s multitude of ad hoc statistics. The only true solution is start with statistics-free, raw data analysis. But how??
When we recently presented posters on our discovery of precision analysis, some of the world’s best-known researchers were literally speechless over its simplicity.
With accurate m/z data, each data-point represents one specific ion. Therefore, akin to using raw light to hunt exoplanets (2019 Physics Nobel), we use raw ions to implicate proteins and their state. In both sciences, the challenge is not fancy statistics but mining voluminous data for evidence of elusive objects.
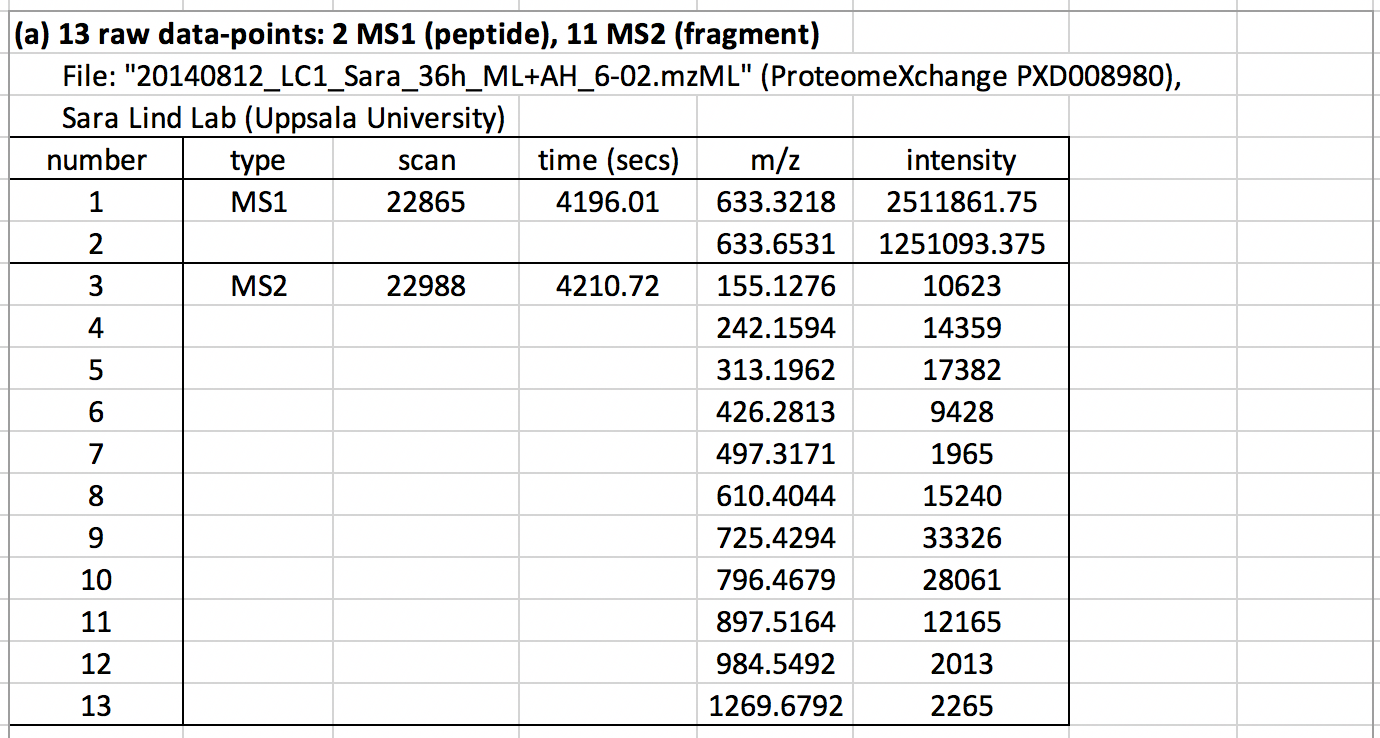
To appreciate the power of precision analysis, consider this challenge: Deduce the experiment and results from just 13 raw MS data-points (Table 1a) from a real-world dataset.
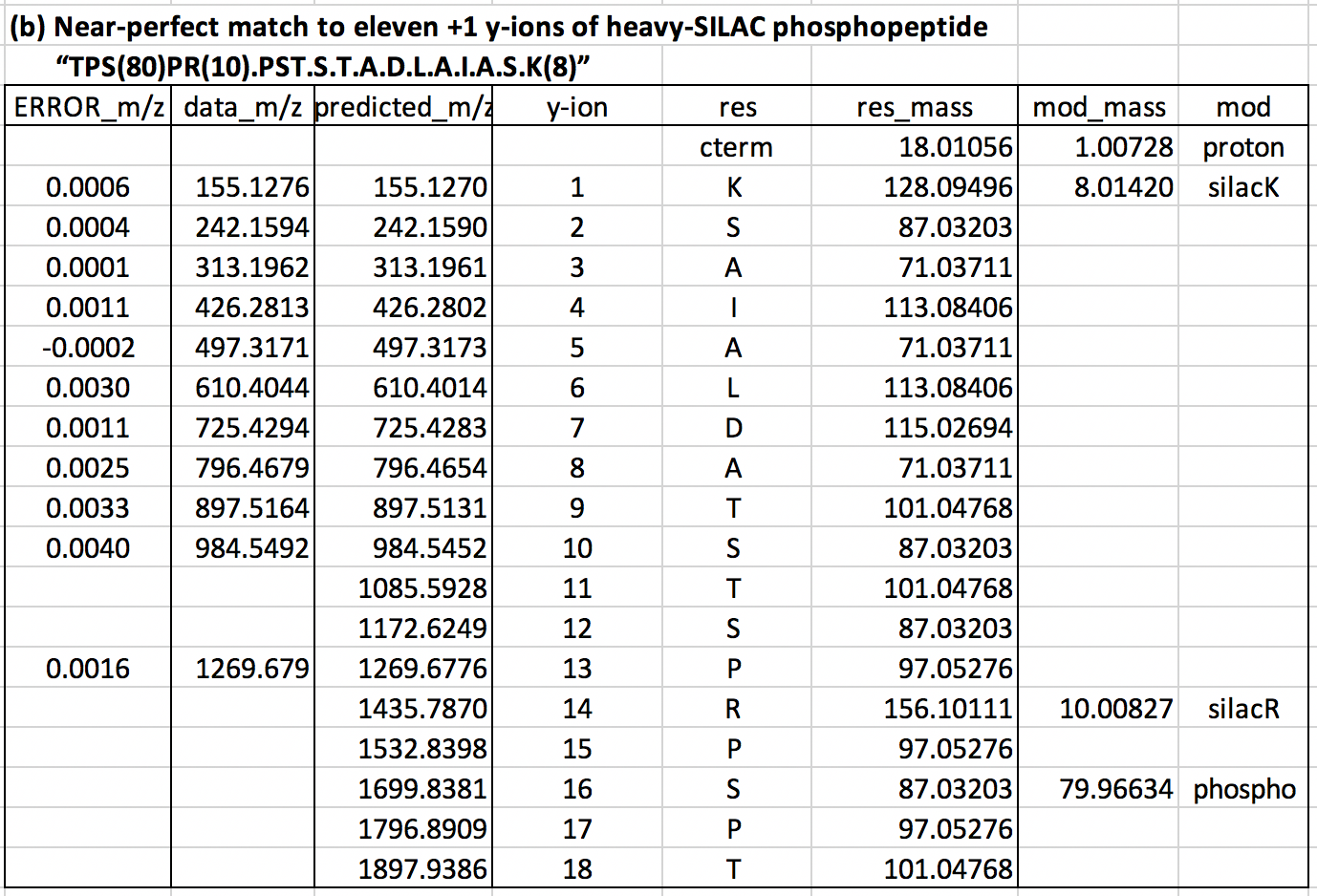
Table 1b below shows your likely analysis.
You would’ve found the MS2 ions are a near-perfect match for the first 10 y-ions (y1 to y10) for the heavy-SILAC peptide “…STADLAIASK”. This pattern appears to be unique to a human adenovirus 2 protein (DNB2_ABE02).
In other words, from just ten MS2 ions (actually 8 are enough), we deduce that: (1) the experiment is likely a SILAC experiment of adenovirus-infected human cells, and (2) the identity of the protein and species.
You can download the Excel data file for Tables 1a & 1b here.
From two MS1 ions showing charge +3, we deduce the peptide is almost certainly the single- phosphorylation heavy-SILAC peptide “TPSPRPSTSTADLAIASK”. The near-perfect y13 fit means the phosphorylation is either T or S beyond y13. Gross quantitation is given the apex MS1 intensity of 1.25M.
Like the Copernican revolution, Precision Proteomics is a fundamentally new paradigm with profound implications. It explains irreproducibility (wrong ions selection), imprecision (statistics overuse), and the futility of PC workflows for “real” research.
The above shows how easy it is to infer from 10+ raw data-points the identity/quantity of a modified protein form (as a representative peptide) including many localized modifications using only arithmetic.
But like a crossword puzzle, it’s easy to confirm a candidate “word” (peptide) is the answer; the challenge is to guess the word from clues in the first place. Here, for just one presumed peptide, we mined ~10 ions from 28M raw ions (24M MS1, 3.6M MS2) in a single 5-year-old data file.
For all modified peptides in all interesting proteins in clinical discovery, we need large-scale data mining of explosive data that requires specialized tech infrastructure ill-suited for inexperienced tech engineers. (For comparison, the closest commercial product may be the “data warehouse” used by large retailers to mine customer profiles for targeted ads, which are typically specialized in-house server systems due to performance, cost, and security vs. cloud computing.)
As a practical matter, peer-review more often than not blocks new ideas especially coming from outsiders. If it were my own research, I would invest in a parallel Precision Proteomics workflow, both to double-check my standard workflow and to learn the new paradigm. Unlike true statistical sciences like genomics, in protein research you just need the right answer before anyone else.
See the posterherefor background information on this peptide example.
1 Comment
Leave your reply.