| Was this article helpful? | |
As a tech veteran among academics, I often feel like a chef among nutritional scientists who equate culinary mastery with learning the most recipes. Like cooking, math and engineering are more art than science, which is why experts can be young with minimal education. While science seeks to distill a complex world into lower dimensional knowns, art finds success within high dimensional unknowns. Here I offer my tech perspective to research success.
In proteomics, the science of analyzing clean data is easy but unimportant. Its true power lies in characterizing ground-breaking one-in-a-million peptides with poor signal-to-noise — a challenge in both engineering and science. Akin to unknowns from discovering Jupiter moons, this cannot be done with beginner PC apps, the fast food of software.
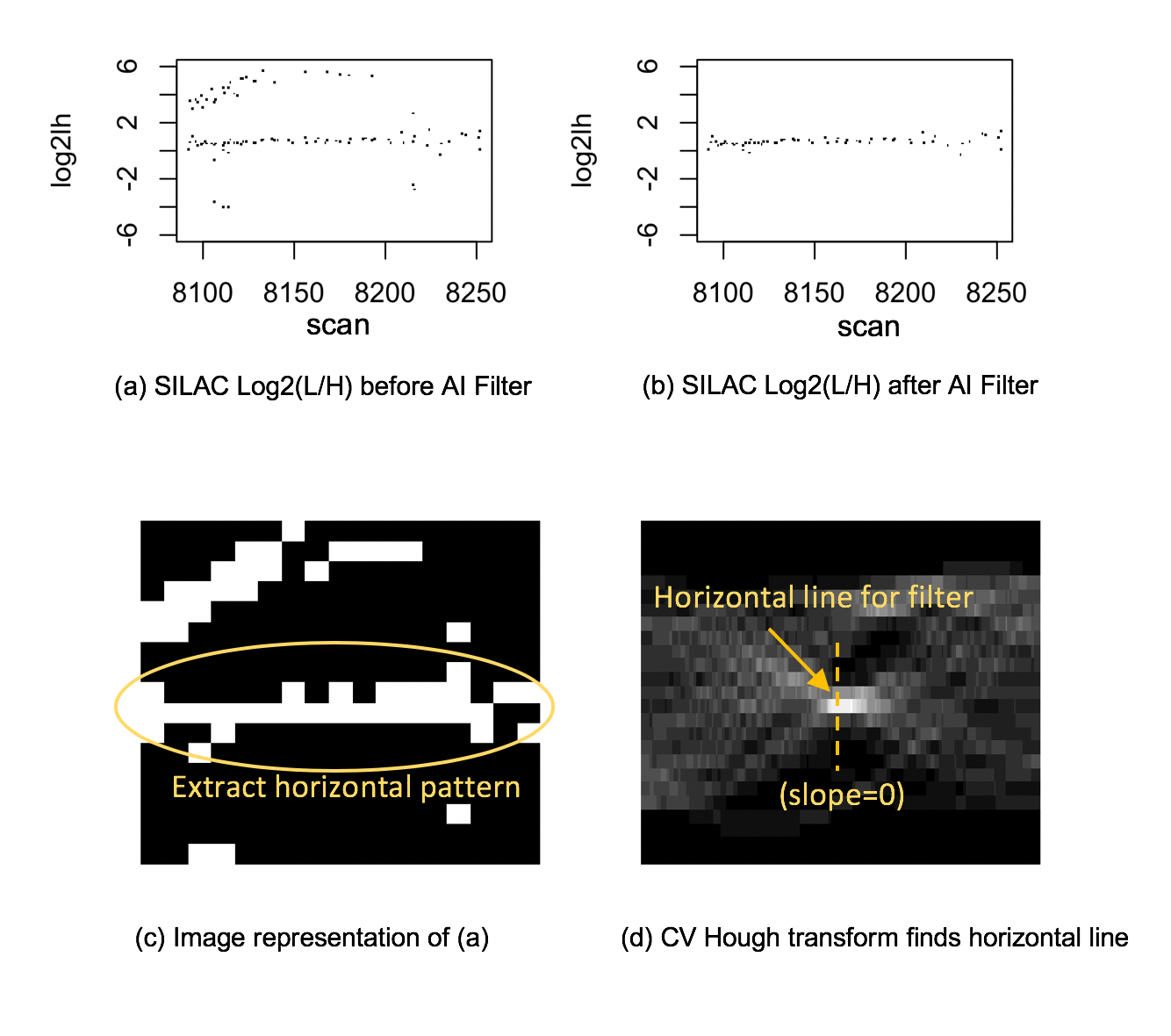
Figure 1: SorcererSILAC uses AI to improve the Light/Heavy estimation. The original mined data-points (a) are filtered using Computer Vision (b). It is converted to an image (c) from which the Hough transform detects a horizontal region of quality data-points. (See ASMS poster for graph details.)
We recently published an ASMS poster with a major breakthrough: For accurate opportune data, as few as 7 fragment ions and about 100 intact peptide ions can be fully analyzed using simple arithmetic — i.e. no statistical models — to identify the peptide/protein with localized chemical modifications.
In other words, with good clean data (majority of most datasets we’ve seen), peptide/protein IDs can be 100% data-driven without cumulative modeling artifacts from using multiple statistical algorithms like PeptideProphet, ProteinProphet, Ascore, and MaxQuant.
Ironically, this brings data analysis back full circle to the early days when the John Yates Lab: (1) used a search engine to gross-guess candidate peptide IDs, (2) used simple software like DTA-Select to discriminate the correct peptide ID as well as gross-identify the protein in a text file, and (3) used simple calculations like area-under-curve to quantify peptides in a second text file. Each result was directly traceable back to raw data — practically impossible with today’s statistical approaches.
This means proteomics just became a precise hard science like astronomy.
It marks a paradigm shift from an imprecise statistical science struggling with irreproducibility. Like all tech-driven fields, the proven roadmap to success is to exponentially maximize productivity with powerful IT to mine hidden treasures before rivals. Since PC apps play no role in any deep information science we know of, they are unlikely to play any role in advancing proteomics despite growing popularity. Indeed, since they rely on aggressive models for speed, their misfit models are likely the source of reputed irreproducible results.
Astronomy offers an insightful analogy to understand mass spec challenges. Faint objects can be identified but not necessarily quantified. Overlapping objects cause ambiguity. And sometimes the data you seek is not captured. They apply equally to proteomics.
We can intuit that, to find a new Jupiter moon, an astronomer may hypothesize at least 3 co-linear dots in timed images, code library functions from “computer vision” (CV), a branch of AI, to mine for them in hi-res images, then summarize possible sightings for detailed expert analysis.
In contrast, no serious astronomer would blindly run a canned PC app that identifies generic objects at 1% error because: (1) PCs lack CPU power to clean noisy data, (2) 1% error is too loose to identify individual objects among millions, and (3) scripting flexibility is needed to explore unknowns.
Putting everything together, the only true solution becomes clear: Find a scalable data mining platform (think MS Excel on steroids) and never buy another software application again. This is possible because all mass spec data are essentially {(m/z, time, intensity)}, plus “ccs” for ion mobility, that’s it. Different applications (e.g. DIA, ETD, different quants) are just different ways to combine them into patterns that scientists should transparently control.
In contrast, canned software have two fundamental problems for deep research. First, they all hardcode assumptions certain to be wrong for some data, causing unpredictably wrong answers. Second, they distance the scientist from raw data which weakens the science. (Prior to proteomics, I’ve not heard of any expert scientist using canned analysis software. It’s always Excel or some other platform they can customize.)
The SORCERER AIMS (AI for MS) is specifically engineered for flexible and scalable proteomics data mining using R, Python, or other Linux languages. The same AIMS app would generally run on all SORCERER systems from the entry system to the largest private cloud instances.
An AIMS app typically uses the modular “cyber-assay” formulation by defining: (1) how MS1 and MS2 raw data-points are mined within time-slices of raw data, (2) what calculations are done on said data within each slice, and (3) how slices are combined into global summaries. The AIMS platform takes care of job queues and scaling to available CPU cores.
The first AIMS app is SorcererSILAC™ for AI-assisted data mining of phospho SILAC datasets. Its main output consists simply of two CSV files for peptide/protein IDs and their quantitation.
Custom AIMS apps can be developed by academic technologists, lab bioinformaticians, or by Sage-N Research on contract. New methodologies such as for novel chemistries or difficult PTMs can be quickly prototyped.
Please contact sales@SageNResearch.com for information and pricing.
Leave a Reply
Send Us Your Thoughts On This Post.