Chemical separation requires multiple steps. Using just one single step means low purity, scarce output, or both. Same with digital separation.
False discovery rate (FDR), the statistical workhorse of proteomics, is widely used (and misused) as a single-step metric to “purify” search engine results into peptide IDs.
Common 1% FDR workflows, designed for academic statistical molecular profiling, are ill-suited for discovery of individual biomarkers. Two problems: way too many false positives while likely omitting low-abundance peptides of clinical relevance. That’s why discovery success proves elusive.
For example, the low-specificity prostate cancer biomarker PSA is said to have abundance beyond the top 1000 proteins. This means biomarker discovery requires accuracy of better than 1 in >100K, or four orders of magnitude better than 1%. So, when you run an experiment and the 1% FDR workflow reports 100K peptide IDs, ~1000 are wrong AND the elusive marker (even if captured in the data) is unlikely to be among them.
The new paradigm of Precision Proteomics [see eNews archivehere] makes data analysis simple (math) but not easy (data mining). With ID/quant of a protein form largely defined by only a dozen raw data-points (like a Saturn moon can be defined by 3 time-elapsed dots), biomarker discovery means efficiently mining millions or trillions of raw ions. Here, FDR is a useful gross pre-filter to narrow the search for semi-interactive analysis.
In our previous eNews example [here], we pretended to study adenovirus proteins in infected human cells. We started with 1% FDR using number and average error of fragment matches (“S-score”) and quickly identified/quantified a adenovirus DNA-binding protein using 13 raw ions. But if we didn’t find what we need, we can try filtering virus peptides to 5% or 10% FDR. (We can easily script a second-step filter, but that’s beyond this email post.)
Note our confident virus peptide ID was based on matching the first ten +1 fragment y-ions to ~0.003 tolerance at a ~15 ppm peptide mass error. Any MS expert can acknowledge this is about as certain as the data allows. In contrast, with conventional 1% FDR workflows, about 1% are statistically certain to be wrong. Since pathogen peptides can never approach 1% of total (or the host is long dead), any such ID is statistically untrustworthy.
FDR for experts
Unlike abstract genes, peptides are real objects with an absolute (if unknowable) identity. (Analogously, whether any telescope dot is really a Saturn moon is absolute if unknowable.) Therefore, FDR of a collection of IDs is also absolute but unknowable. In other words, any calculated FDR is merely an estimate that can be no more accurate than ~1%. But the error can be far higher depending on statistical rigor.
FDR is commonly estimated using “target-decoy” search whereby real protein sequences (target) are augmented with reversed sequences (decoy). Since they are roughly 50:50 distributed, it’s commonly assumed that any wrong ID is 50:50 distributed as target-vs-decoy.
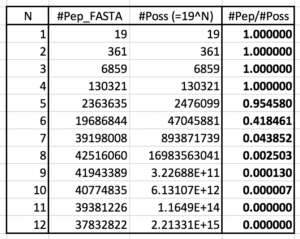
It turns out that’s only asymptotically true starting at length 8 and higher. Almost all possible peptides of length 5 or less are found in nature (see table).
Therefore, all short search results are 100% target regardless of whether correct or wrong. Mathematically, this means any calculated FDR value is really a lower bound, not a true estimate. This is due to palindrome sequences as well as any sequence whose reversed form happens to also be a forward sequence, which includes all short peptides.
In other words, the FDR is a useful but nuanced and often-misleading statistic. Precision Proteomics shows it is best used as a gross pre-filter instead of deriving the main output.
The SORCERER script to compute all possible peptides is available for download [fasta2mer_sorc].
Deep data analysis requires tech experts. Statistical profiling can be forgiving, but precise biomarker data mining is full of minefields. Consider bringing in Silicon Valley tech professionals to complement your team when precise answers matter.
Leave a Reply
Send Us Your Thoughts On This Post.